Photochemistry of 2,6-Dicyano-N,N,N',N'-tetramethyl-p-phenylenediamine.

Photochemistry of 2,6-Dicyano-N,N,N',N'-tetramethyl-p-phenylenediamine. | |
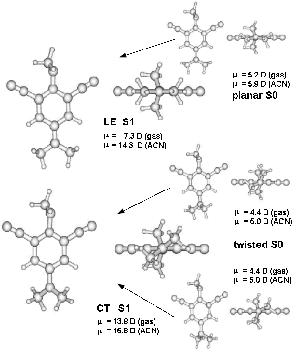
Conclusion:
CASSCF calculations of the title compound
result in three ground state conformations A, B and C with
different orientations of the amino-methyl groups on one side of the aromatic
ring (structures on the right side in the figure). Conformers
B and C are less stable than A (top right structure)
by more than 8 kJ/mol, therefore only A can
be seen in experimental investigations like NMR, IR spectra.
During excitation, the amino group on one side of the ring
rotates (in B and C) and
becomes planar (in all conformers)
resulting in only two excited state geometries A* and C* (structures
on the left side in the figure).
This is accompanied by a raise of the dipole moment.
Whereas in the gas phase only C* shows a large dipole moment, in the solvent
acetonitril both excited state conformers A* and C* have
a very large dipole moment. Such a large dipole moment
change can also be seen in the
experimental data estimating an excited state dipole moment of around 11 D
from the solvatochromic shift.
The experimental Stokes shift is in good agreement with
the computed difference between the absorption and emission energies.
This large dipole moment change is associated to a large geometry change
resulting in an antiquinoid structure with planar amino group in A* and a
twisted amino group in C* and to a charge transfer from the NMe2 groups
to the aromatic system.
Summarizing the computational results and comparing them to the experimental
data, A* represents the locally excited state (LE) and C* is clearly
the charge transfer state (CT).
The reason why no dual fluorescence can be observed in the experimental
flourescence spectra, can
be explained by the energetics of the excited state: conformer C* lies by more
than 8 eV higher on the energy scale than A*, and the solvation does not
bring the LE and CT states closer together. Therefore conformer A* is
the only detected emitting state. Its antiquinoid geometry causes its
charge transfer character, which is the reason for the measured Stokes shift.