Dr. Michael Ramek: Research.
 deutsche Version.
deutsche Version.Dr. Michael Ramek: Research. |
deutsche Version. |
Main research activity is the structure determination of biologically important compounds via quantum chemical ab initio methods.
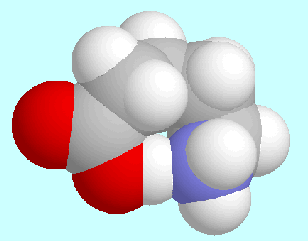 For several
years the focus had been on intramolecular hydrogen bonds
in amino- and hydroxy compounds (FWF-projects
P6856 (1988-1990),
P8053 (1990-1991), and
P9095 (1992-1994));
the results of these studies have
found application, e.g., in the field of shape-similarity
analysis in cooperation with Prof. Paul G. Mezey (University of Saskatchewan,
Saskatoon, Canada). In the recent years the main targets are
derivatives of indole-3-acetic acid
and model peptides (see below).
For several
years the focus had been on intramolecular hydrogen bonds
in amino- and hydroxy compounds (FWF-projects
P6856 (1988-1990),
P8053 (1990-1991), and
P9095 (1992-1994));
the results of these studies have
found application, e.g., in the field of shape-similarity
analysis in cooperation with Prof. Paul G. Mezey (University of Saskatchewan,
Saskatoon, Canada). In the recent years the main targets are
derivatives of indole-3-acetic acid
and model peptides (see below).
 For a number of years the symmetrization of wavefunctions and
quantum states has been investigated in
a series of publications, most of them in cooperation with
Prof. Bruno Gruber(Southern Illinois University,
Carbondale, IL (USA)).
For a number of years the symmetrization of wavefunctions and
quantum states has been investigated in
a series of publications, most of them in cooperation with
Prof. Bruno Gruber(Southern Illinois University,
Carbondale, IL (USA)).
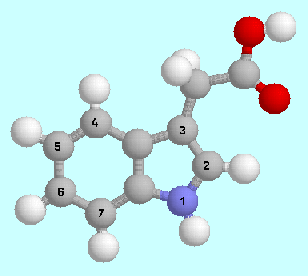 Indole-3-acetic acid
and its derivatives are growth hormones in plants (auxins),
which govern many biological processes such as cell divisions and
enlargement, developmental differentiation, and the syntheses of
specific proteins. In a
series of papers, co-authored by Dr. Sanja Tomić
(Institut Ruđer Bošković, Zagreb, Croatia),
it could be shown that the potential energy surfaces of these compounds are
very sensitive to substitution at a specific position of the indole
nucleus (position 4), whereas substitution by ethyl or chlorine at
the other positions of the phenyl ring has no influence on the potential energy
surfaces. Although the form of the potential energy surface cannot
be directly correlated with biological activity, because other factors
(lipophilicity or correlation with other compounds like cytokinines)
are also of importance, these studies give important clues
to the forces that determine auxin activity.
Indole-3-acetic acid
and its derivatives are growth hormones in plants (auxins),
which govern many biological processes such as cell divisions and
enlargement, developmental differentiation, and the syntheses of
specific proteins. In a
series of papers, co-authored by Dr. Sanja Tomić
(Institut Ruđer Bošković, Zagreb, Croatia),
it could be shown that the potential energy surfaces of these compounds are
very sensitive to substitution at a specific position of the indole
nucleus (position 4), whereas substitution by ethyl or chlorine at
the other positions of the phenyl ring has no influence on the potential energy
surfaces. Although the form of the potential energy surface cannot
be directly correlated with biological activity, because other factors
(lipophilicity or correlation with other compounds like cytokinines)
are also of importance, these studies give important clues
to the forces that determine auxin activity.
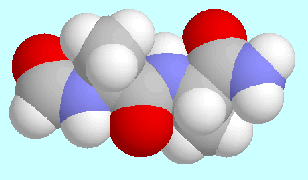 Peptides
are mostly modeled by attaching a formyl or acetyl group
to the N-terminus of a short peptide chain and by converting the C-terminus
to an amide group at the same time.
The peptide chain may as small as a single residue X,
in which case the model compound
Peptides
are mostly modeled by attaching a formyl or acetyl group
to the N-terminus of a short peptide chain and by converting the C-terminus
to an amide group at the same time.
The peptide chain may as small as a single residue X,
in which case the model compound