Structure Determination of Auxin Phytohormones. |
|
| T1 | T2 | E [kJ/mol]
|
|---|
| 0.000° |
0.000° |
0.000
|
| 112.508° |
103.570° |
0.499
|
| 99.061° |
-96.409° |
1.998
|
| 111.850° |
1.601° |
4.361
|
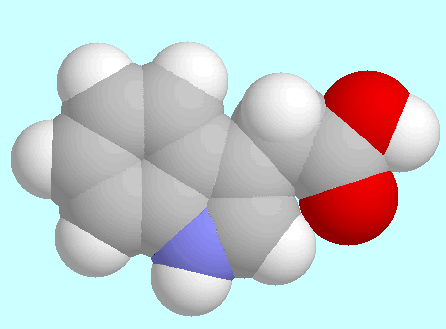
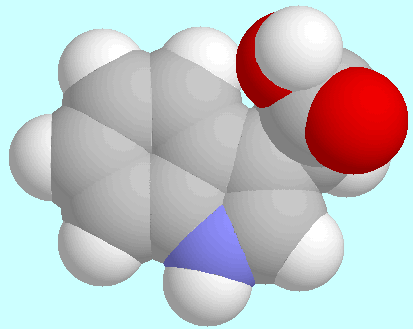
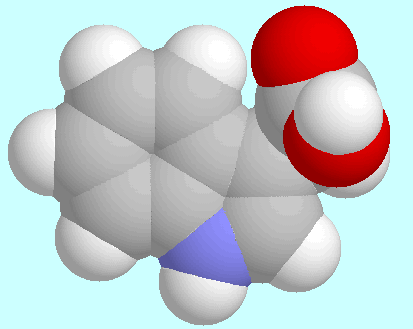
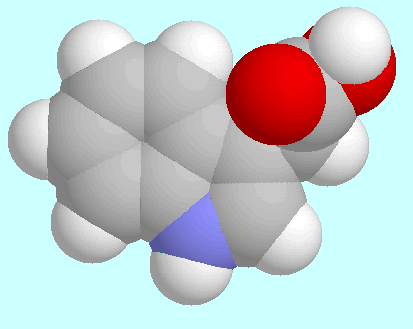
The potential energy surface of 3-indole acetic acid (IAA)
contains four symmetry unique local minima with the COOH group in
cis-orientation.
The geometries of these conformers are shown below this paragraph in
the order of their energy.
The global minimum is mirror symmetrical and stabilized by a weak
C=O···H-C2 hydrogen bond;
the other three conformers all exhibit
a tilted side chain, with three different orientations of the
COOH group. The 6-31G*-optimized positions in
T1/T2-space and the corresponding
relative energies are collected in the table next to this paragraph.
A number of basis sets, ranging from STO-3G to 6-311G** have been
employed to optimize these conformers at RHF-level. The comparison
of the calculated rotational constants with the experimental values
shows that the 4-31G values are already within the experimental error
of 1%; in contrast, the electron densities
indicate that one set of d-polarization functions is necessary
to obtain an adequate description of the aromatic ring.
The ab initio results compare in an interesting manner with
those of molecular mechanics (MM) calculations, which were performed by Tomić
et al. with a number of different
force fields: none of the force fields used was able to reproduce the
ab initio
results; the best agreement was obtained with CVFF and MM3.
All of the MM calculations gave only two symmetry
unique minima, which correspond to the ab initio
conformers of second and third energy, but failed to reproduce the two other
conformations as energy minima. The planar conformation, which is the
global minimum in the ab initio case, is described as
a second order saddle point by MM methods.

 introduction.
introduction.
© publications
(our own and selected others).
 4-Cl-IAA.
4-Cl-IAA.
 Quantum Chemistry Group.
Quantum Chemistry Group.
Informations required by Austrian law
(Offenlegung gem. §25 MedienG): Dr. Michael Ramek, Graz.