Ching-Hsing Yu, Lothar Schäfer, and
Michael Ramek,
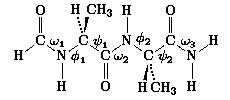
Local Geometry Trends and Torsional Sensitivity in
N-Formyl-L-alanyl-L-alanine Amide
and the Limitations of the Dipeptide Approximation
J. Phys. Chem. A, 103, 8337-8345 (1999).

 ,
,
 ,
,