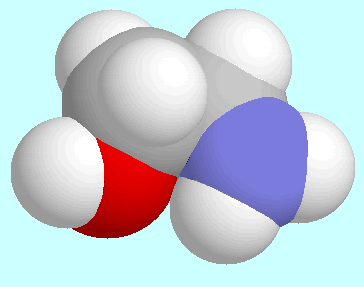
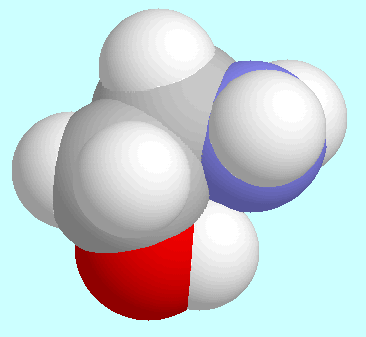
2-Aminoethanol can form two different interactions between
the two functional groups: O-H···N and
O···H-N. There is one symmetry-unique
conformer with the former interaction, which is the global minimum of
the potential energy surface. The geometry of this conformer, which is
shown next to this paragraph, is of the envelope-type: the
O-H···N-C
sequence is almost co-planar, whereas the remaining carbon atom (C1)
is located out of that plane. The interaction O-H···N
is a hydrogen bond,
according to all of the usual criteria, and it stabilizes this conformer
(and its enantiomer) thermodynamically as well as kinetically:
the conformer, which is second lowest in energy, has a relative
energy of approximately 7 kJ/mol, and the lowest potential barrier,
which happens to lead to this very conformer, is around
10.5 kJ/mol.
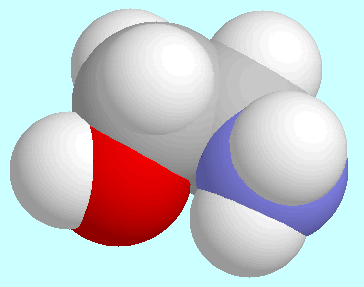
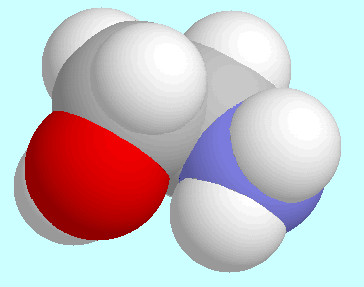
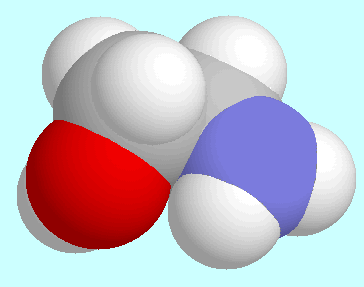
The O···H-N-interaction is present
in four symmetry-unique conformers,
which are shown below this paragraph. The
O···H-N-interaction is
not a hydrogen bond: the bond order is less than 0.01, the N-H bond lengths
and vibrations frequencies do not deviate from the mean value of the
other conformers, and there is no saddle point in the electron density
between the atoms O and H. However, there is an electrostatic interaction,
which prevents the formation of a third orientation of the amino group; it
also causes these conformers to be slightly lower in energy than most of the
other (extended)
conformers, namely second, third, fifth, and sixth in energy.


 2-Aminoethanol can form two different interactions between
the two functional groups: O-H···N and
O···H-N. There is one symmetry-unique
conformer with the former interaction, which is the global minimum of
the potential energy surface. The geometry of this conformer, which is
shown next to this paragraph, is of the envelope-type: the
O-H···N-C
sequence is almost co-planar, whereas the remaining carbon atom (C1)
is located out of that plane. The interaction O-H···N
is a hydrogen bond,
according to all of the usual criteria, and it stabilizes this conformer
(and its enantiomer) thermodynamically as well as kinetically:
the conformer, which is second lowest in energy, has a relative
energy of approximately 7 kJ/mol, and the lowest potential barrier,
which happens to lead to this very conformer, is around
10.5 kJ/mol.
2-Aminoethanol can form two different interactions between
the two functional groups: O-H···N and
O···H-N. There is one symmetry-unique
conformer with the former interaction, which is the global minimum of
the potential energy surface. The geometry of this conformer, which is
shown next to this paragraph, is of the envelope-type: the
O-H···N-C
sequence is almost co-planar, whereas the remaining carbon atom (C1)
is located out of that plane. The interaction O-H···N
is a hydrogen bond,
according to all of the usual criteria, and it stabilizes this conformer
(and its enantiomer) thermodynamically as well as kinetically:
the conformer, which is second lowest in energy, has a relative
energy of approximately 7 kJ/mol, and the lowest potential barrier,
which happens to lead to this very conformer, is around
10.5 kJ/mol.