The potential energy surface of
3-aminopropanionamide contains a total of
six symmetry-unique conformers, which are shown below this paragraph
in the order of their energy.
This is an astonishing small number, since the "general wisdom" of
organic structure chemistry predicts three possible orientations
for internal rotations of the C-N and C-C bonds;
for HN-CH-CH-CONH
this would mean 3³ = 27 local minima, i.e., 14 symmetry-unique conformers. One might
expect that the difference between 14 conformers-to-be-expected and the six,
which are actually observed, is caused by a number of strong
intramolecular interactions.
This is not the case, however: there are only two intramolecular
interactions, namely N···H-N(CO),
which is found in the global
minimum, and N-H···O=C,
which is present in the conformers of second
the third lowest energy.
The hydrogen bond N···H-N(CO) in the global minimum
has a H···N distance of 2.107 Å
and a bond order of 0.056.
This hydrogen bond is rather weak, according to all indicators (e.g.,
the hydrogen bond length is 78% of the sum of the van der Waals radii,
and the H-N bond length increase is only 0.006 Å).
Furthermore, the relative energies of all six conformers are
more or less equally distributed in the range between 0 and 19.1 kJ/mol,
which also indicates no major stabilization from this hydrogen bond.
The second intramolecular interaction, N-H···O=C,
is no hydrogen bond
according to all of the usual criteria.
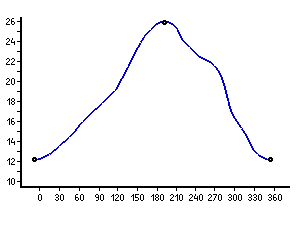
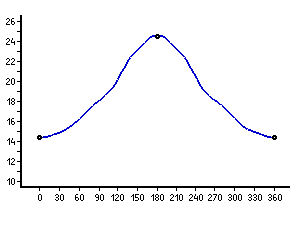
One of the mysteries of this potential energy surface with so few
local minima is the internal rotation of the CONH-group in
the extended conformers (fourth and fifth in energy): in both cases
the energy increases steadily up to a transition state,
in which this group is rotated by 180°; after passing
the transition state, the path leads back to
the original orientation. Slight shoulders appear in these energy
profiles (see below), but without any tendency towards stationary points.
Energy profile of the internal rotation of the CONH-group
in the extended forms with asymmetric (left or top) and
symmetric (right or bottom)
orientation of the amino group. Values are given in kJ/mol (relative to
the global minimum) and degree (C-C-C=O), stationary points are marked.


 N-CH
N-CH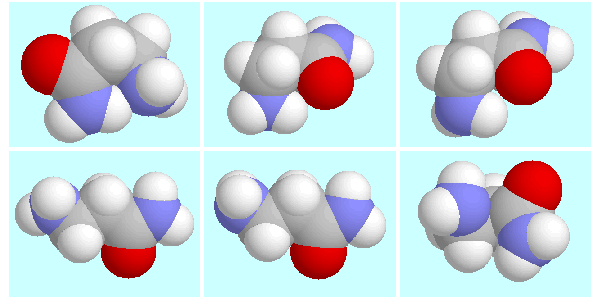
 -alanine.
-alanine.